


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 D) insertion into the C-C bond in cyclopropane and ethane
D) insertion into the C-C bond in cyclopropane and ethane黒崎 譲; 高柳 敏幸
Chemical Physics Letters, 355(5-6), p.424 - 430, 2002/04
被引用回数:3 パーセンタイル:8.65(Chemistry, Physical)シクロプロパンのC-C結合に対するO(D)挿入反応の入り口付近における5つの最低一重項ポテンシャルエネルギー面を、CASPT2/cc-pVDZレベルで計算した。その結果、5枚のポテンシャル面の内の最も下にあるものは、入り口付近で引力的であるのに対し、他の4枚は斥力的であることが予測された。比較のため、エタンについて同様の計算を行った結果、5枚のポテンシャル面は入り口付近ですべて斥力的であることが予測された。これらの計算結果は、O(D)とアルカン分子の反応についての最近の実験結果と矛盾しない。
 H
H +Cl reaction in water; Ab initio molecular orbital study using a cluster model
+Cl reaction in water; Ab initio molecular orbital study using a cluster model黒崎 譲
Journal of Physical Chemistry A, 105(49), p.11080 - 11087, 2001/12
被引用回数:3 パーセンタイル:9.19(Chemistry, Physical)CH+Cl反応の水中における電荷分離過程を、反応系に4個の水分子を加えたクラスターモデルを用いて非経験的分子軌道法により研究した。その結果、最終生成物である電荷分離錯体(M3)は反応系の解離極限、CH+Cl+4HO、より4.3kcal/molエネルギー的に低く、反応系で生成する錯体(M1)より0.6kcal/mol高いことが予測された。また、気相中の自由エネルギーを求め、それに溶媒和自由エネルギーを加えることによって全自由エネルギーを計算したところ、M3の全自由エネルギーはM1より5.1kcal/mol低いことが予測された。この結果は水中におけるこの反応系の電荷分離過程が自発的であることを強く示唆する。
+CH H+CH reaction and its isotopic variants
H+CH reaction and its isotopic variants黒崎 譲; 高柳 敏幸
Journal of Chemical Physics, 113(10), p.4060 - 4072, 2000/09
被引用回数:22 パーセンタイル:55.85(Chemistry, Physical)反応H+CHH+CH(1)及びこれを同位体置換した反応、HD+CHH+CHD(2), DH+CHD+CH(3),D+CHD+CHD(4),H+CDH+CHD(5)の反応速度定数を、トンネル補正を加えた変分的遷移状態理論により計算した。その結果、これらの反応に見られる同位体効果はほとんど一次同位体効果によるもので、二次同位体効果及び反応経路(IRC)の曲率の効果は比較的小さいことが明らかとなった。このことは、分子軌道計算からも明らかなように、これらの反応のポテンシャルが「early」であることに起因すると思われる。また、反応1と2の反応速度定数の計算結果は、実験結果とかなり良い一致を示した。
NO( B
B )NO(
)NO(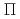 )+H reaction
)+H reaction黒崎 譲*; 高柳 敏幸
Journal of Molecular Structure; THEOCHEM, 507(1-3), p.119 - 126, 2000/07
反応HNO(2 )NO()+Hの機構について非経験的分子軌道法を用いて理論的に研究した。基底状態と第一励起状態のポテンシャル面を適当な2つの内部座標の関数として、FOCI/cc-PVTZレベルで計算した。その結果、HNO(B)の分子面に垂直なCs面を保持した反応経路上に、conical intersectionが存在することが明らかとなった。同じ反応経路上には遷移状態が存在することも、既に明らかであったが、電子波動関数の対称性から、この遷移状態を経由する反応は対称禁制であることが予測されていた。しかし、今回の計算結果から、反応系はconical intersectionを回避する形で対称性をCsからCに落とすことにより禁制を解くということが明らかとなった。
)NO()+Hの機構について非経験的分子軌道法を用いて理論的に研究した。基底状態と第一励起状態のポテンシャル面を適当な2つの内部座標の関数として、FOCI/cc-PVTZレベルで計算した。その結果、HNO(B)の分子面に垂直なCs面を保持した反応経路上に、conical intersectionが存在することが明らかとなった。同じ反応経路上には遷移状態が存在することも、既に明らかであったが、電子波動関数の対称性から、この遷移状態を経由する反応は対称禁制であることが予測されていた。しかし、今回の計算結果から、反応系はconical intersectionを回避する形で対称性をCsからCに落とすことにより禁制を解くということが明らかとなった。
H+ClCHCl reaction黒崎 譲*
Journal of Molecular Structure; THEOCHEM, 503(3), p.231 - 240, 2000/05
本研究では、まず気相中における反応CH+ClCHClの機構について理論的に検討した。極限的反応座標(IRC)計算の結果、得られた反応物、遷移状態(TS)、生成物が1つの反応経路上にあることが確認された。反応の活性化エネルギーはPMP4及びB3LYPレベルで、それぞれ36.3,35.9kcal/molと計算された。次に、溶媒中における同反応の機構について検討した。その結果、極性溶媒中では気相中のようなTSが存在しないことが予測された。また、気相中では励起状態であったCHCl +Cl
+Cl が、極性溶媒中では基底状態となることが明らかとなった。誘電率80の極性溶媒中では、CHCl+Clのエネルギー値は反応物(CH+Cl)と比較して11.2kcal/molとなることがB3LYPレベルで計算された。このことは、この反応が気相中よりも極性溶媒中でより起こりやすいことを示唆している。
が、極性溶媒中では基底状態となることが明らかとなった。誘電率80の極性溶媒中では、CHCl+Clのエネルギー値は反応物(CH+Cl)と比較して11.2kcal/molとなることがB3LYPレベルで計算された。このことは、この反応が気相中よりも極性溶媒中でより起こりやすいことを示唆している。
 +HCH+H and CD+HCDH+H reactions using variational transition state theory and the multidimensional semiclassical tunneling method
+HCH+H and CD+HCDH+H reactions using variational transition state theory and the multidimensional semiclassical tunneling method黒崎 譲*; 高柳 敏幸
Journal of Chemical Physics, 110(22), p.10830 - 10842, 1999/06
被引用回数:20 パーセンタイル:53.61(Chemistry, Physical)反応CH+HCH+H(I)及びCD+HCDH+H(II)の反応速度における同位体効果について、変分的遷移状態理論及び準古典的多次元トンネリング法を用いて理論的に考察した。まず、反応IとIIのポテンシャル面を量子化学的手法により計算した。次に、得られたポテンシャル面を用いて、多次元トンネリングを準古典的に考察した変分的遷移状態理論により反応速度定数を求めた。実験的には、5Kの固体パラ水素中で、反応IIの方が反応Iより反応速度が速いことが報告されている。ここでの計算の結果、理論的にも反応IIの方が反応Iよりも5Kで反応速度が速いことが予測され、実験結果を定性的に説明することができた。
+HCH+H reaction黒崎 譲*; 高柳 敏幸
Chemical Physics Letters, 299(1), p.57 - 63, 1999/00
被引用回数:15 パーセンタイル:43.56(Chemistry, Physical)反応CH+HCH+H(I)及びCD+HCDH+H(II)における異常な同位体効果について理論的に考察した。実験的には、固体パラ水素中でCHI(CDI)を光分解し、CH(CD)を生成させて5Kでしばらく放置すると、CHI/p-H系ではCHの生成は確認されなかったがCDI/p-H系ではCDHの生成が確認された。すなわち、反応Iは起こらないが反応IIは起こることが見出された。本理論計算では、この同位体効果を説明するために、反応I,IIの固有反応座標(IRC)を高精度の非経験的分子軌道法により求め、さらにIRCに直交する基準振動の振動数も計算し、反応途中でのゼロ点振動エネルギーの値も見積もった。その結果、従来の意味での同位体効果は予想通りほとんどないが、ゼロ点振動エネルギーを考慮したeffectiveなポテンシャルを比較すると、反応IIの方が反応Iより反応障壁が低く障壁の幅も小さいことが明らかとなった。このことは反応IIの方がトンネル確率が大きいことを意味しており、上の実験結果を良く説明している。
SHS+SH reaction黒崎 譲; 高柳 敏幸
Journal of Chemical Physics, 111(23), p.10529 - 10536, 1999/00
被引用回数:13 パーセンタイル:39(Chemistry, Physical)反応H+HSHS+SHの速度定数を半古典多次元トンネリングによる補正を加えた変分的遷移状態理論により、100-2500Kの温度範囲で計算した。計算結果は広い温度範囲で実験値と一致した。特に室温付近での計算値と実験値の一致は極めて良好であった。本研究では、非経験的分子軌道計算によって得られたポテンシャルエネルギーを全く調整することなしに、反応速度の非アーレニウス的挙動を定量的に再現することができた。
D)+HO reaction黒崎 譲*; 高柳 敏幸
Journal of Physical Chemistry A, 103(3), p.436 - 442, 1999/00
被引用回数:20 パーセンタイル:53.61(Chemistry, Physical)反応N(D)+HOについて、非経験的分子軌道法を用いて反応機構を考察した。この結果、おもな最終生成物はNH(
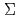 )+OH()及びHNO(A')+Hであることが予測された。この結果は、過去の高精度の計算結果ならびに最近の実験結果と一致している。また、反応の初期段階として、NのO原子への付加、NのOH結合への挿入、NによるH原子の引き抜き、の3つの機構が考えられるが、このうちNの付加が最も有利な機構であることが計算により示された。
)+OH()及びHNO(A')+Hであることが予測された。この結果は、過去の高精度の計算結果ならびに最近の実験結果と一致している。また、反応の初期段階として、NのO原子への付加、NのOH結合への挿入、NによるH原子の引き抜き、の3つの機構が考えられるが、このうちNの付加が最も有利な機構であることが計算により示された。
高橋 節子*; 鈴谷 賢太郎; 小原 真司*; 小浦 延幸*; L.A.Curtiss*; Saboungi, M. L.*
Zeitschrift f r Physikalische Chemie, 209, p.209 - 221, 1999/00
r Physikalische Chemie, 209, p.209 - 221, 1999/00
AlCl-1-エチル-3メチル-イミダゾリウムクラロイドは、室温型の溶融塩として大変有望な系であり、多くの研究がなされているが、構造に関しては大変複雑な系であるので研究が少なくよくわかっていない。そこで、本研究では、この系の46~67mol%融液について中性子回折によってその構造を調べた。また、回折結果を理解するために、融液中での存在が認められているAlCl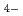 ,AlCl
,AlCl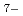 ,EMIイオンについて非経験的分子軌道計算を行い、各イオンが独立の場合と陽陰イオン間の相互作用を取り入れた場合についてそれぞれのイオンの構造を調べた。その結果、中性子回折の結果は分子軌道計算による短範囲構造(イオンの構造)をよく反映しており、特にイオン間相互作用を取り入れた場合の構造がよく一致することが明らかになった。
,EMIイオンについて非経験的分子軌道計算を行い、各イオンが独立の場合と陽陰イオン間の相互作用を取り入れた場合についてそれぞれのイオンの構造を調べた。その結果、中性子回折の結果は分子軌道計算による短範囲構造(イオンの構造)をよく反映しており、特にイオン間相互作用を取り入れた場合の構造がよく一致することが明らかになった。
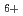 , H
, H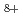 , H
, H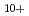 , H
, H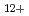 and H
and H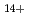
黒崎 譲*; 高柳 敏幸
Journal of Chemical Physics, 109(11), p.4327 - 4334, 1998/09
被引用回数:19 パーセンタイル:52.68(Chemistry, Physical)非経験的分子軌道法を用いて、偶数個の水素原子より成る水素クラスターカチオン(H,H,H,H,H)の構成とエネルギーを求めた。水素クラスターカチオンの生成実験においては、奇数クラスターのみが生成するものと従来から考えられていたが、比較的最近になって偶数クラスターが副生成物として検出された。クラスターの構造は、奇数、偶数いずれの場合も、3角形のH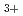 が核となってそのまわりにHやHが弱く結合しているものが考えられていたが、Hクラスターの場合にH
が核となってそのまわりにHやHが弱く結合しているものが考えられていたが、Hクラスターの場合にH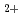 が核となる構造の方がHが核となるものよりもわずかにエネルギー的に安定になるという計算が発表された。今回は、Hについても同様のことを示すと同時に、H核のHクラスターまでを計算し、少なくともHまでは安定に存在し得ることを示した。この結果はHよりも大きな偶数クラスターを検出できなかったというKirchner&Bowersの実験結果と定性的に一致した。
が核となる構造の方がHが核となるものよりもわずかにエネルギー的に安定になるという計算が発表された。今回は、Hについても同様のことを示すと同時に、H核のHクラスターまでを計算し、少なくともHまでは安定に存在し得ることを示した。この結果はHよりも大きな偶数クラスターを検出できなかったというKirchner&Bowersの実験結果と定性的に一致した。
cluster. An ab initio molecular orbital study黒崎 譲*; 高柳 敏幸
Chemical Physics Letters, 293(1-2), p.59 - 64, 1998/00
被引用回数:19 パーセンタイル:52.68(Chemistry, Physical)Hクラスターの異性化反応の機構について、非経験的分子軌道法を用いて考察した。構造最適化はMP2/cc-pVTZレベルで行い、1点エネルギー計算はQCISD(T)/cc-pVTZレベルで行った。Hクラスターには、対称性がCs及びDdの2つの異性体があることが既に理論的に予測されていたが、今回、異性化反応の遷移状態(TS)が理論的に見い出された。極限的反応座標(IRC)計算によって、このTSが2つの異性体を結ぶ異性化反応経路上の鞍点に位置することが確認された。H(CS)クラスターから見た相対的エネルギー値は、TSが+0.4kcal/mol,H(Dd)クラスターが-4.0kcal/molであることがQCISD(T)レベルで予測された。
elimination from 2,3-dimethylbutane cation: (CH)CHCH(CH)(CH)C=C(CH)+H; An ab initio molecular orbital study黒崎 譲*; 高柳 敏幸; 宮崎 哲郎*
Journal of Molecular Structure; THEOCHEM, 452, p.209 - 218, 1998/00
2,3-ジメチルブタンカチオン((CH)CHCH(CH),h-DMB)からのH脱離反応に対し、非経験的分子軌道計算を行った。構造最適化はUMP2/6-31G(d)レベルで行い、1点エネルギー計算をUMP3/6-31G(d)及びUMP4(SDTQ)/6-31G(d)レベルで行った。その結果、この反応は障壁が22-24kcal/molで26-29kcal/mol発熱的であることが予測された。非経験的分子軌道計算から得られたデータを用い、遷移状態理論に基づいて量子力学的(トンネル)効果を考慮した熱反応速度定数を求めると、h-DMBの反応の速度定数は77Kで約10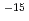 s
s と予測された。h-DMBにおいて、脱離するHをDで置換したカチオン(d-DMB)の反応の速度定数は77Kで約10
と予測された。h-DMBにおいて、脱離するHをDで置換したカチオン(d-DMB)の反応の速度定数は77Kで約10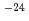 sと計算された。このことから、h-DMBからのH脱離反応にはトンネル効果が重要であることが示唆される。一方、h-DMBの反応速度定数に対する実測値は約12桁も大きい。これは量子化学計算のレベルがまだ低いことを示唆する。
sと計算された。このことから、h-DMBからのH脱離反応にはトンネル効果が重要であることが示唆される。一方、h-DMBの反応速度定数に対する実測値は約12桁も大きい。これは量子化学計算のレベルがまだ低いことを示唆する。
城戸 健太朗; 笠原 健人*; 佐藤 啓文*; 横川 大輔*
no journal, ,
分子性液体論に基づく3次元溶媒和理論(MC-MOZ法)は、溶液内の化学過程を理解する上で鍵となる自由エネルギープロファイルや溶媒和構造を分子レベルで評価する強力な方法論である。しかし、化学結合の解離と生成を伴う過程への適用は難しかった。本研究では、MC-MOZ法と標準的な第一原理分子軌道法との結合によってこの点を解決し、より広範な溶液内化学過程に応用可能な新規方法論を開発した。この新規方法論はQM/MM法と同等の物理量を与え、溶液のマルチフィジックスモデルの一つと位置付けられる。水溶液内の水分子やホルムアルデヒド分子、及びSN2反応へ適用した。関連した方法との比較を行い、その有効性を議論した。
